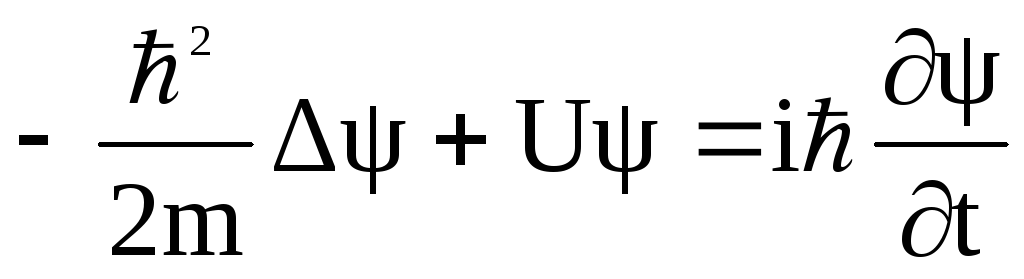
Anisotropic Hole Transport in a p-Quaterphenyl Molecular Crystal: Theory and Simulation
By A. Ya Freidzon, A. A. Bagaturyants, Ya. V. Burdakov, V. R. Nikitenko, and V. A. Postnikov
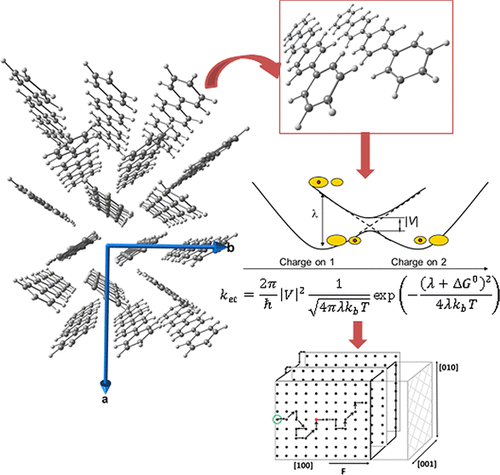
A computational procedure is proposed for predicting the charge hopping rate in organic semiconductor crystals. The procedure is verified using a p-quaterphenyl molecular crystal as the test system, in which the thermally activated hole mobility is relatively low, its hole states are localized, and, hence, charge transport is of hopping character. The hole mobility in p-quaterphenyl is simulated by the Monte Carlo method with the hopping probability governed by a Marcus-like rate constant. The microscopic parameters of the Marcus model have been calculated by ab initio multireference quantum chemical method (XMCQDPT/CASSCF). Molecular conformation and crystal environment effects on the Marcus hopping parameters are studied. It is found that different arrangements of monomers typical for the crystal structure provide different hopping parameters and, hence, different hole mobilities in different directions. Monte Carlo simulations of the hole mobility predict that the hole mobility attains its maximum in the [100] direction, where hopping occurs through parallel monomers at the closest distance, which is lower than 0.01 cm2/(V·s).