Effective Fragment Potential Method for H-Bonding: How To Obtain Parameters for Nonrigid Fragments
by Nikita Dubinets and Lyudmila V. Slipchenko.
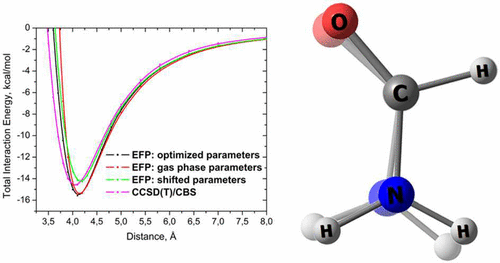
Accuracy of the effective fragment potential (EFP) method was explored for describing intermolecular interaction energies in three dimers with strong H-bonded interactions, formic acid, formamide, and formamidine dimers, which are a part of HBC6 database of noncovalent interactions. Monomer geometries in these dimers change significantly as a function of intermonomer separation. Several EFP schemes were considered, in which fragment parameters were prepared for a fragment in its gas-phase geometry or recomputed for each unique fragment geometry. Additionally, a scheme in which gas-phase fragment parameters are shifted according to relaxed fragment geometries is introduced and tested. EFP data are compared against the coupled cluster with single, double, and perturbative triple excitations (CCSD(T)) method in a complete basis set (CBS) and the symmetry adapted perturbation theory (SAPT). All considered EFP schemes provide a good agreement with CCSD(T)/CBS for binding energies at equilibrium separations, with discrepancies not exceeding 2 kcal/mol. However, only the schemes that utilize relaxed fragment geometries remain qualitatively correct at shorter than equilibrium intermolecular distances. The EFP scheme with shifted parameters behaves quantitatively similar to the scheme in which parameters are recomputed for each monomer geometry and thus is recommended as a computationally efficient approach for large-scale EFP simulations of flexible systems.